

|
|||||
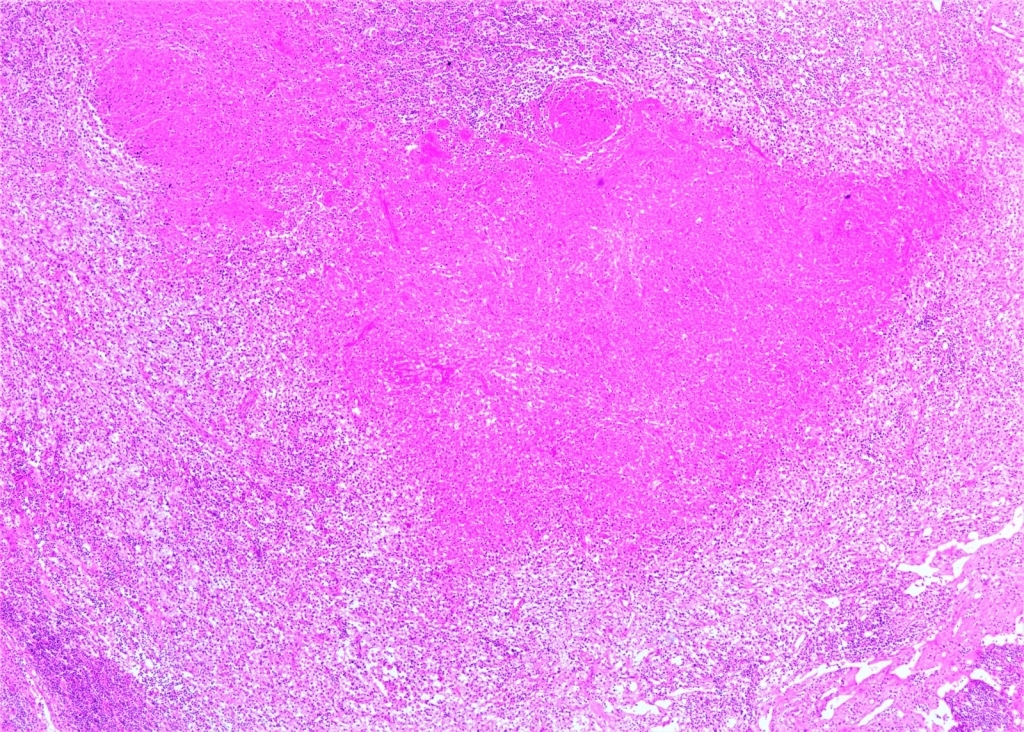
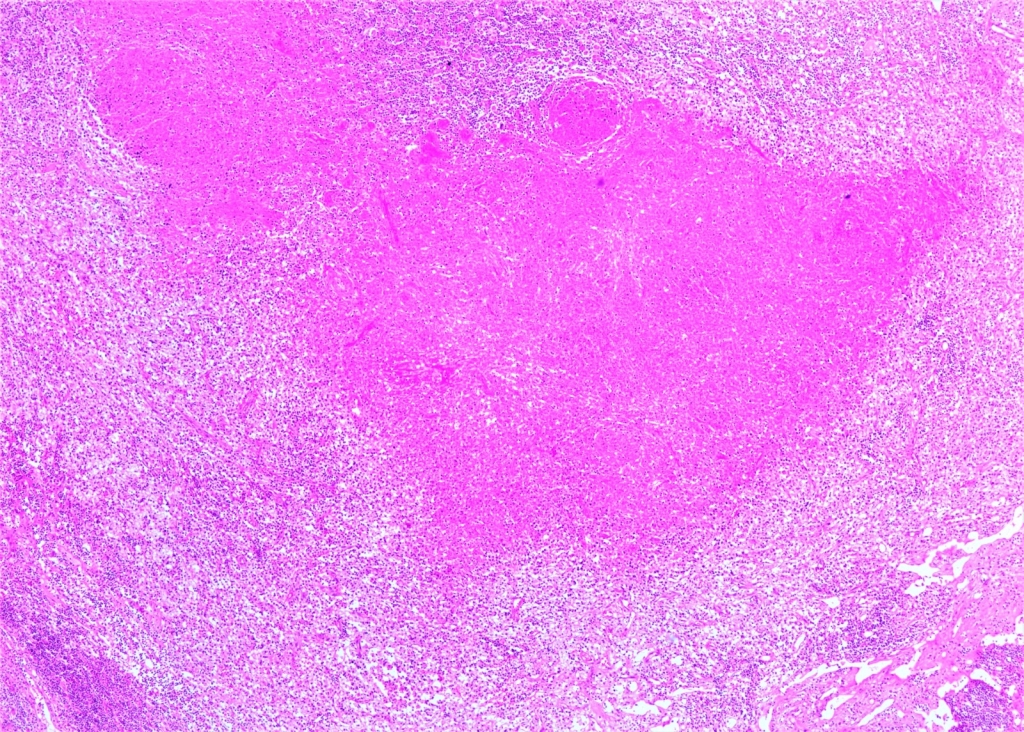
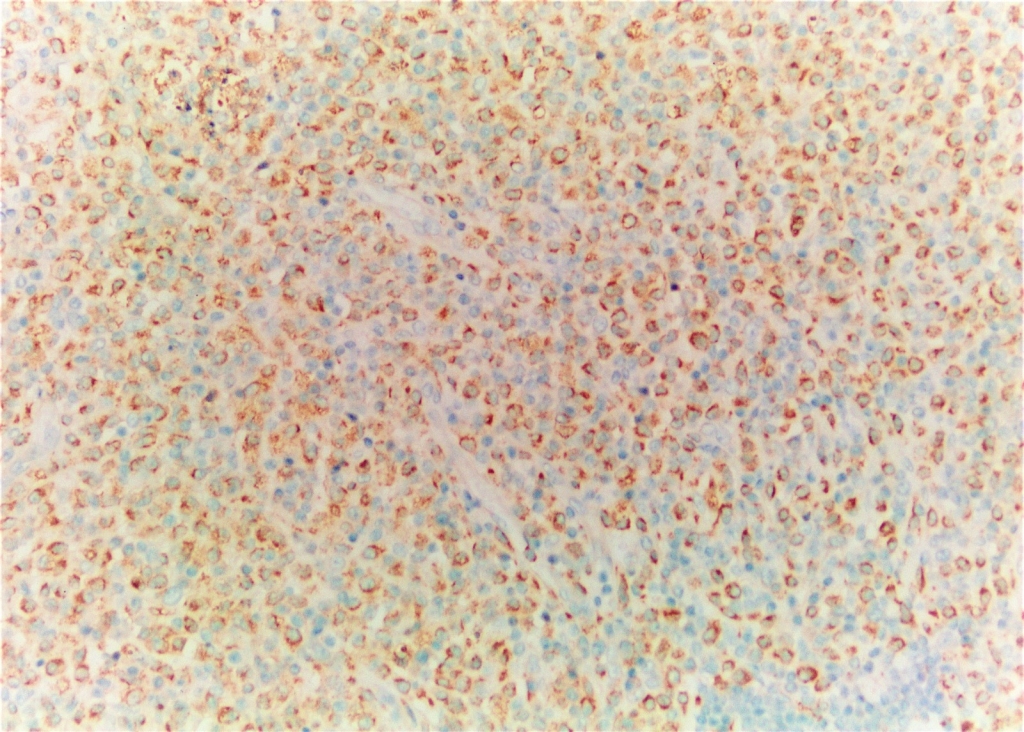
AbstractKikuchi–Fujimoto Disease (KFD) is a rare condition. Due to its rarity, accurate diagnosis is often delayed, and patients may receive incorrect diagnoses and treatments. Moreover, this disease may precede the development of various systemic illnesses. Early diagnosis allows for a more controlled disease course. In this study, we present two patients who were admitted to our clinic with complaints of cervical swelling and were diagnosed with KFD based on excisional biopsy findings.IntroductionKikuchi–Fujimoto Disease (KFD), also known as histiocytic necrotizing lymphadenitis, was first described in Japan in 1972 by Kikuchi and Fujimoto. This rare disease is typically a self-limiting inflammatory condition. However, a few reports in the literature have documented fatal outcomes. Although KFD is thought to predominantly affect children and young adults of Asian descent, cases have been reported in individuals of various ages and ethnic backgrounds. KFD usually presents with an acute or subacute clinical course. In addition to systemic symptoms such as fever, fatigue, weight loss, arthralgia, and skin lesions, the disease is characterized by painful, tender, and mobile cervical lymphadenopathy (LAP) [1,2] . Most patients exhibit relative leukocytosis, leukopenia, or neutropenia. Since there is no specific test for the disease, definitive diagnosis is made through histopathological examination of an excisional lymph node biopsy. However, some studies have suggested that fine-needle aspiration biopsy may also be helpful in establishing the diagnosis [3]. In this report, we present two patients who presented to our clinic with complaints of neck swelling and were diagnosed with KFD after thorough evaluation. Through these cases, we aim to raise clinicians’ awareness of this rare condition. Case ReportCASE 1 A 42-year-old male patient presented to our clinic with a one-month history of painful swelling in the neck. On physical examination, a mobile, tender, approximately 3 cm mass was palpated at the left cervical level 2A. Other routine ear, nose, and throat (ENT) examinations were unremarkable. Neck ultrasonography (USG) revealed an asymmetrical lymph node with a thickened cortex measuring 33 × 11 mm, located deep to the left sternocleidomastoid (SCM) muscle near the mandibular angle, and showing central vascularity. Laboratory investigations showed a borderline elevated erythrocyte sedimentation rate (ESR: 22). Other routine blood tests were within normal limits. Viral screenings for Epstein–Barr virus (EBV), Herpes Simplex virus (HSV), and Cytomegalovirus were all negative. An excisional biopsy was planned. The LAP was excised from the left cervical level 2A. Histopathological examination confirmed the diagnosis of KFD. Histopathological evaluation demonstrated extensive geographic necrosis within the lymph node in the absence of neutrophilic infiltration, a characteristic finding of KFD ( Figure 1,2).
CASE 2 An 18-year-old female patient presented to our clinic with complaints of neck swelling, night sweats, and a weight loss of approximately 10 kg over the past year. On physical examination, a palpable mass measuring approximately 2 cm was detected in the right cervical levels 3–4. Other routine ENT examinations were unremarkable. Neck USG showed multiple lymph nodes in both cervicojugular regions, the largest being 23 × 11 mm in the right cervical level 2 and 20 × 7.5 mm in the left cervical level 2. These lymph nodes had thick cortices and non-visible fatty hilum. Neck magnetic resonance imaging (MRI) revealed multiple fusiform LAPs, the largest measuring 23 × 10 mm on the right and 22 × 8 mm on the left. Laboratory tests were mostly normal except for mild anemia (Hemoglobin: 11.1 g/dL). Specific testing revealed EBV and antinuclear antibody positivity. An excisional biopsy was planned. The LAP was excised from the posterior aspect of the right SCM. Histopathological examination confirmed the diagnosis of KFD. DiscussionKFD is a rare condition with an unknown specific incidence. Although cases have been reported worldwide across all ethnic groups, it is more commonly observed in Japanese and other Asian populations. The disease primarily affects adults under the age of 40, but it has also been documented in patients ranging from 19 months to 75 years. It was previously thought to be more common in females; however, recent studies suggest that both sexes are equally affected [4]. Potential etiological factors include EBV and human herpesvirus types 6–8, but many studies argue against a direct pathogenic role. In our report, one of the patients tested positive for EBV. KFD typically presents with acute or subacute onset and usually resolves within 2 to 3 weeks. Cervical LAP is present in 56–98% of cases, typically in the posterior cervical triangle. Painful LAP occurs in up to 59% of patients, while generalized LAP is reported in 1–22% of cases. Mild fever and upper respiratory tract symptoms occur in 30–50% of patients with KFD. In our report, one patient had isolated LAP, while the other also experienced weight loss and night sweats. Extranodal involvement is rare and typically affects the skin, but may occasionally involve bone marrow or the liver [5-7]. In our cases, no extranodal involvement was observed. Cutaneous involvement most frequently affects the face and upper body and can manifest as nodules, erythematous or indurated papules, erythema multiforme, or maculopapular lesions [8,9]. No skin lesions were observed in our patients. Most patients have normal laboratory results. Mild anemia, elevated ESR, and increased C-reactive protein (CRP) may be seen in some cases. Other findings may include leukopenia (especially granulocytopenia) and leukocytosis. Atypical lymphocytes in the peripheral blood have been reported in about one-third of patients [10]. In our cases, one had elevated ESR and the other had mild anemia, with no other significant lab abnormalities. Imaging modalities such as USG, Computed Tomography (CT) , and MRI can be utilized in KFD. In one case involving neurological symptoms, MRI was used for imaging [11]. Another study highlighted the common CT findings in KFD and suggested that these findings may help in diagnosis and follow-up [12]. However, there is no definitive imaging modality to distinguish KFD from malignancy. Due to the non-specific clinical, laboratory, and imaging features, KFD can be easily mistaken for lymphoma, particularly in early-stage LAP without prominent necrosis. One study reported that 18F-fluorodeoxyglucose positron emission tomography (PET) may aid in differentiating KFD from lymphoma [13]. Definitive diagnosis of KFD is established through histopathological examination. However, even histopathological interpretations can sometimes be misleading. In one study, a patient was initially diagnosed with lymphoma and started on chemotherapy, but a repeat histological review revealed KFD, leading to cessation of treatment. The patient’s symptoms resolved within 5 months [14]. Despite the diagnostic challenges at presentation, the prognosis of KFD is generally favorable. Although the incidence of systemic lupus erythematosus developing in the context of KFD appears low, it has been reported in 2 of 61 patients in one study and in 4 of 169 patients in another [15]. For this reason, follow-up by a rheumatology clinic is recommended. ConclusionKFD is a benign and self-limiting condition characterized by localized LAP. However, it can easily be confused with other diseases. Histopathological confirmation is crucial to avoid misdiagnosis and inappropriate treatment. In both of our cases, early surgical excision and histopathological analysis allowed for accurate diagnosis without delay, ensuring proper clinical follow-up. AcknowledgementDeğerli Editörlerimiz, "Kikuchi-Fujimoto Hastalığı: İki Olgu Sunumu" başlıklı yazımızı ENTcase dergisinde yayınlanmak üzere değerlendirmenize sunmaktan mutluluk duyuyorum. Bu olgu sunumu, nadir görülen ve sıklıkla yanlış teşhis edilen bir hastalık olan Kikuchi-Fujimoto hastalığının iki örneğini sunarak, kulak burun boğaz alanındaki klinik özelliklerini ve tanı zorluklarını vurgulamaktadır. Bu raporun klinisyenler için ilgi çekici olacağına ve literatüre değerli bilgiler sağlayacağına inanıyoruz. Bu makale özgündür, daha önce yayınlanmamıştır ve başka bir yerde yayınlanması için değerlendirme aşamasında değildir. Tüm yazarlar makalenin son halini inceleyip onaylamış ve hazırlanmasına önemli katkılarda bulunmuşlardır. Bu gönderimle ilgili herhangi bir çıkar çatışması olmadığını beyan ederiz. Makalemizin hakem değerlendirmesi ve saygıdeğer derginizde olası bir yayın için değerlendirilmesini saygılarımızla rica ederiz. Daha fazla bilgi veya belgeye ihtiyacınız olursa lütfen benimle iletişime geçmekten çekinmeyin. Zamanınız ve ilginiz için teşekkür ederim. Saygılarımla Informed Consentolgu sunumu yaptığımız iki hastadan.References
|
|||||
| Keywords : Kikuchi – Fujimoto hastalığı;Histiyositik Nekrotizan Lenfadenit;Servikal Lenfadenopati;Ayırıcı Tanı | |||||
|